Quel est le processus menant à l’autorisation des médicaments biosimilaires?
Le parcours des médicaments biosimilaires jusqu’à l’autorisation est raccourci
Fresenius Kabi est conscient de l’importance des médicaments biosimilaires et s’engage en faveur d’un traitement efficace et sûr des patients. Pour pouvoir être autorisés, les médicaments biosimilaires doivent passer avec succès une série de tests stricts de la FDA et de l’EMA. Les similarités spécifiques avec le produit de référence doivent être démontrées par les moyens suivants:
Études analytiques: Démonstration que les médicaments biosimilaires présentent des propriétés moléculaires et fonctionnelles très similaires à celles du produit de référence autorisé.
Études cliniques: Confirmation de l’efficacité, de la tolérance, de la pharmacocinétique et de la pharmacodynamique équivalentes par comparaison directe avec le produit de référence chez une population de patients appropriée et dans une indication sensible.
Développement du médicament biosimilaire
Démonstration que le médicament biosimilaire est très similaire au produit de référence.
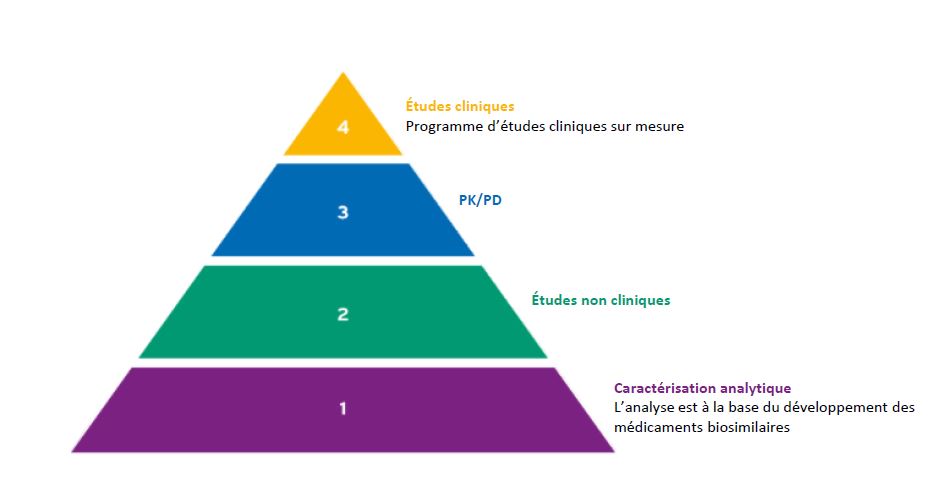
Le développement des médicaments biosimilaires met l’accent sur la caractérisation et l’analyse moléculaires. Un programme d’études cliniques sur mesure permet de confirmer la biosimilarité.
Le parcours des médicaments biosimilaires jusqu’à l’autorisation est raccourci. Une fois qu’il a été démontré qu’un médicament biosimilaire est très similaire au produit de référence, il est alors scientifiquement justifié de s’appuyer, dans le cadre du processus d’autorisation, sur certaines connaissances et expériences existantes concernant la sécurité et l’efficacité du produit de référence.
Le processus de développement raccourci permet un programme de développement de médicaments potentiellement plus rapide et moins coûteux pour un médicament biosimilaire.
"Les données obtenues au cours de dix années d’expérience clinique montrent que les médicaments biosimilaires approuvés par l’EMA peuvent être utilisés, pour toutes les indications approuvées, de manière aussi sûre et efficace que les autres médicaments biologiques" (citation de l’EMA)
"Tous les produits biologiques approuvés par la FDA, y compris les produits de référence et les produits biosimilaires, font l’objet d’une évaluation rigoureuse afin de rassurer les patients sur l’efficacité, la sécurité et la qualité de ces produits" (citation de la FDA)
Autorisation de médicaments biosimilaires
Le premier médicament biosimilaire a été autorisé par la Commission européenne en 2006. Pour être autorisés dans l’UE, les médicaments biologiques et biosimilaires doivent se soumettre à une procédure centrale très réglementée et contrôlée via l’EMA. L’examen par l’EMA donne lieu à un avis scientifique qui est ensuite transmis à la Commission européenne, laquelle peut finalement délivrer une autorisation de mise sur le marché à l’échelle de l’UE. En Suisse, c’est Swissmedic, l’institut des produits thérapeutiques et l’autorité suisse d’autorisation des médicaments, qui, comme pour les génériques, est responsable de l’autorisation des médicaments biosimilaires. Le premier médicament biosimilaire en Suisse a été autorisé en 2009.
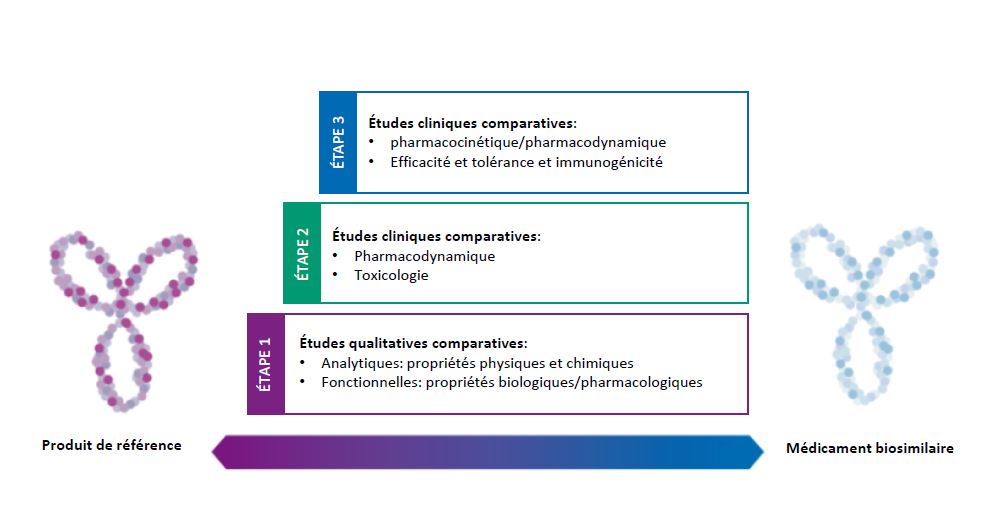
Dans le cadre de la procédure d’autorisation de mise sur le marché, le développement des médicaments biosimilaires s’appuie essentiellement sur des études comparatives directes pour établir la biosimilarité avec le produit de référence. Pour cela, le médicament biosimilaire et le produit de référence sont directement comparés au cours d’un processus en plusieurs étapes adapté à chaque produit: Les premières études comparatives de la qualité (étape 1) permettent de déterminer l’étendue et le type des études non cliniques (étape 2) et cliniques (étape 3) nécessaires à l’étape suivante du développement, afin d’exclure les différences potentielles entre le médicament biosimilaire proposé et le produit de référence.
.
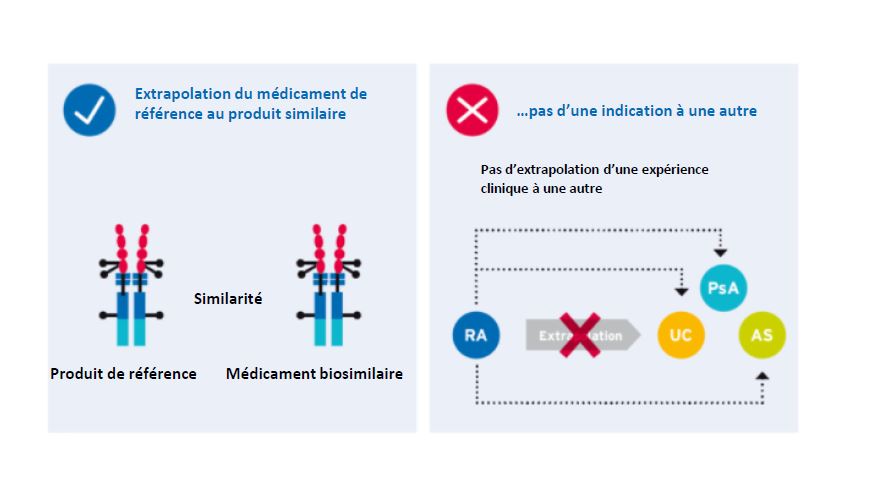
Médicaments biosimilaires et concept de l’extrapolation
Dans le cadre du processus d’autorisation d’un médicament biosimilaire, l’extrapolation des données d’efficacité et de sécurité à d’autres indications du produit de référence pour lesquelles la comparabilité clinique n’a pas été testée est possible sur la base de l’ensemble des données et d’une justification scientifique appropriée. Ainsi, certaines indications nécessitent moins d’études cliniques avec le médicament biosimilaire, voire aucune, ce qui accélère le processus d’autorisation et permet aux patients d’accéder à plus d’options thérapeutiques. L’extrapolation n’est cependant pas un processus automatique. Elle obéit au contraire à des dispositions strictes. Ainsi, le mécanisme d’action de la substance active doit être le même dans les différentes indications. De plus, l’ensemble des données disponibles sur la caractérisation structurelle et fonctionnelle du médicament biosimilaire et la comparabilité clinique dans une indication sensible avec le produit de référence doit permettre de démontrer de manière scientifiquement concluante que l’on peut supposer une efficacité et une tolérance très similaires, même dans des indications extrapolées. Une molécule fortement similaire se comportera également de manière très similaire dans toutes les indications et tous les groupes de patients.
Extrapolation à d’autres indications pour les biosimilaires